La science derrière le pipeline de l’hémophilie de Sanofi
Les personnes hémophiles sont en attente de traitements à effets plus pérennes leur permettant de mieux gérer la maladie dans leur quotidien. C’est pourquoi nos équipes de R&D étudient actuellement trois approches pour prévenir le saignement chez les personnes présentant des déficits de facteurs de coagulation :
- Fabrication de protéines : dans le cadre de nos études cliniques, nous étudions la possibilité d’élaborer un nouveau type de traitement par facteurs VIII de remplacement à demi-vie prolongée. Celui-ci viserait à maintenir une protection élevée contre les saignements chez les personnes atteintes d’hémophilie A, dont les niveaux de facteurs se situent à l'échelle normale ou quasiment normale pendant une majeure partie de la semaine.
- Technologie de l’ARN : notre objectif est de permettre aux personnes hémophiles de mener une vie active sans devoir être préoccupé en permanence de leurs traitements. C’est pourquoi nous nous efforçons de fournir une protection étendue tout en allégeant la charge de la prise du traitement. Nous étudions la possibilité d’élaborer un traitement à base d’un petit ARN interférent (ARNip). Celui-ci viserait à rééquilibrer les facteurs de coagulation chez les personnes atteintes d’hémophilie A et B, avec ou sans inhibiteurs (anticorps qui s’attaquent aux facteurs de remplacement), et de limiter potentiellement l’administration prophylactique à seulement six injections par an.
- Thérapie génique : nos chercheurs étudient des moyens de corriger la mutation d’ADN unique qui empêche les patients de fabriquer suffisamment de facteurs de coagulation.
1. Thérapies par facteur de remplacement : la fabrication de protéines à l’œuvre
Sans une quantité suffisante de facteurs VIII ou IX, l’organisme ne peut produire suffisamment de thrombine pour activer la coagulation.1,2 Pour pallier à ce problème, de nombreuses personnes sont soignées avec des facteurs de remplacement, administrés par perfusions intraveineuses. Ces facteurs de remplacement circulent dans le sang jusqu’à être métabolisés ou activés pour arrêter un saignement.
Mais les protéines de facteurs sont éliminées si rapidement de l’organisme que de nombreuses personnes atteintes d’hémophilie A nécessitent une perfusion de facteurs de remplacement plusieurs fois par semaine.
Les traitements par facteurs de remplacement à demi-vie prolongée permettent aux protéines de facteurs VIII ou IX de circuler plus longtemps. Et ce, grâce à des innovations antérieures en matière de fabrication de protéines. Pour éviter que les facteurs de remplacement ne soient métabolisés trop rapidement, les scientifiques les ont conçus de manière à ce qu’ils « se fixent » sur des protéines à la durée de vie plus longue.
Dépasser le seuil des 19 heures
Mais il y a une limite : le facteur VIII de coagulation ne peut circuler dans le sang plus de 15 à 19 heures. Et ce, parce que le facteur VIII est généralement accompagné d’une autre protéine, le facteur von Willebrand (vWF), qui empêche son élimination3,4, mais toutes les 15 à 19 heures, la moitié du vWF est éliminé du sang, emportant avec lui le facteur VIII.
Pour les chercheurs, tout l’enjeu réside dans la mise au point d’un traitement par facteurs de remplacement VIII qui maintienne ces protéines de coagulation en circulation pendant plus de 19 heures.
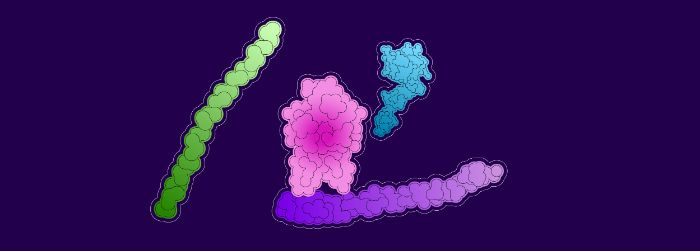
Le facteur von Willebrand (violet) évite que le facteur VIII (rose) ne soit éliminé par certaines protéines (vertes et bleues).
Une alternative au facteur von Willebrand ?
Nous avons développé un facteur VIII de remplacement qui bénéficie d’une protection similaire à celle du vWF, sans être éliminé avec le facteur vWF.5
- Un petit fragment du vWF vient se fixer au facteur de remplacement tel un leurre, afin d’inciter le vWF de l’organisme à l’ignorer. Une molécule « de liaison » libère le leurre lorsque le facteur de remplacement est nécessaire à la coagulation.
- Une autre molécule, XTEN®, fait office de bouclier et repousse les protéines afin d’éviter que le facteur de remplacement ne soit métabolisé. Un domaine d’anticorps permet de maintenir le facteur plus longtemps dans la circulation sanguine.
L’objectif de cette nouvelle solution thérapeutique, encore à l’étude, est de maintenir des niveaux élevés de facteurs VIII de remplacement dans le sang pendant plusieurs jours.
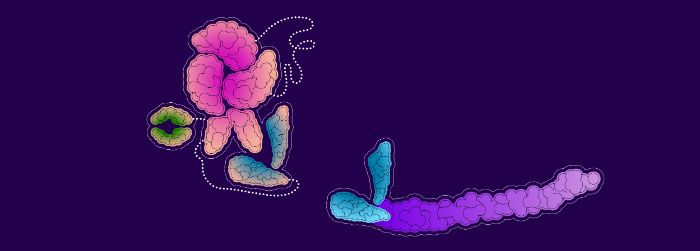
Lorsque le facteur VIII de remplacement (jaune) est lié à un domaine vWF (bleu), le vWF circulant dans le sang (bleu et gris) ne peut pas s’y lier.
2. La technologie de l’ARN dans la lutte contre l’hémophilie
L’ARNm s’est fait connaître pour son rôle dans la vaccination, mais d’autres technologies à ARN peuvent ouvrir de nouvelles possibilités en matière de soins. Nos équipes cherchent notamment à améliorer la coagulation du sang chez les personnes atteintes d’hémophilie en utilisant un petit ARN interférent (ARNip).6,7,8 Celui-ci peut être programmé de manière à ralentir la fabrication par l’organisme de protéines antithrombine III.
L’antithrombine est une protéine qui contribue à la circulation sanguine en limitant la quantité de thrombine produite par l’organisme. Les personnes dont le sang est moins riche en antithrombine produisent plus de thrombine que les autres. Récemment, les scientifiques ont découvert qu’un taux plus faible d’antithrombine III pouvait rétablir la production de thrombine chez les personnes atteintes d’hémophilie, rendant ainsi la coagulation plus efficace.9,10,11
Comment ralentir la production d’antithrombine III ?
L’organisme se base sur des modèles d’ARNm pour fabriquer les protéines d’antithrombine III.12 Sans ces modèles d’ARNm, les protéines ne peuvent être produites. Nos équipes étudient la possibilité d’établir un complexe d’enzymes/ARNip capable d’intercepter les modèles d’ARNm utilisés pour la fabrication d’antithrombine III et d’en éliminer suffisamment pour ralentir la production.13,14 Cela pourrait permettre de maintenir les niveaux d’antithrombine III bas, et ainsi de disposer de davantage de thrombine pour initier la coagulation.
C’est précisément l’idée derrière la mise au point d’un traitement prophylactique potentiel pour les personnes atteintes d’hémophilie A ou B, avec ou sans inhibiteurs.15,16,17 Ce type de solution thérapeutique pourrait simplifier encore les cycles de traitement pour les patients.
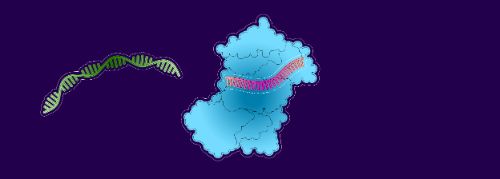
1. L’ARNip attire une molécule d’ARNm destinée à produire des modèles de fabrication de protéines d’antithrombine III
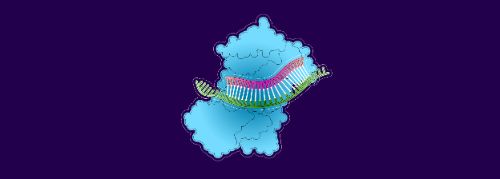
2. L’ARNm se lie à l’ARNip
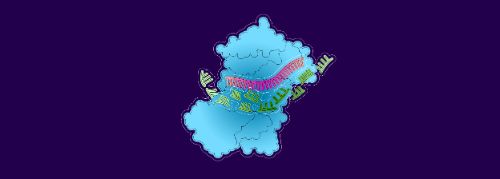
3. Les enzymes détruisent l’ARNm et l’évacuent...
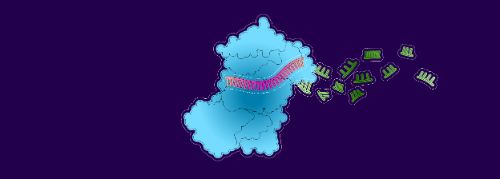
4. ... laissant libre le brin de l’ARNip pour accueillir une autre molécule d’ARNm
3. Thérapies géniques
Pour les chercheurs en hémophilie, l’objectif de la thérapie génique est de soigner la maladie en corrigeant la mutation d’ADN unique qui empêche le patient de fabriquer suffisamment de facteurs VIII ou IX. Nous explorons de nombreuses approches expérimentales en matière de thérapie génique qui, nous l’espérons, amélioreront un jour le quotidien des personnes atteintes d’hémophilie et de leurs proches.18,19
Cet article fait référence à des thérapies actuellement à l’étude dans le cadre de nos programmes de recherche et développement. La sécurité et l’efficacité des médicaments expérimentaux décrits ici n’ont pas encore été évaluées par les autorités réglementaires.
En savoir plus

Pour les héroïnes de l’hémophilie
De nouvelles technologies pour restaurer un bon équilibre sanguin
La R&D au service des troubles hématologiques rares
Références
- Siegemund T, et al. (2003) Thrombin generation in severe haemophilia A and B: the endogenous thrombin potential in platelet-rich plasma. Thromb Haemost 90:781-786; doi:10.1160/TH03-01-0027
- Brummel-Ziedins KE, et al. (2009) Thrombin generation and bleeding in haemophilia A. Haemophilia 15:1118-1125; doi:10.1111/j.1365-2516.2009.01994
- Orlova NA, et al. (2013) Blood Clotting Factor VIII: From Evolution to Therapy. Acta Naturae 5:19-39
- Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124(9):1412-1425. doi:10.1182/blood-2014-05-378638
- Seth Chhabra E, Liu T, Kulman J, et al. (2020) BIVV001, a new class of factor VIII replacement for hemophilia A that is independent of von Willebrand factor in primates and mice. Blood 135:1484-1496; doi: 10.1182/blood.2019001292
- Bartz S, Jackson AL (2005) How will RNAi facilitate drug development? Sci STKE: Signal Transduction Knowledge Environment. 295:pe39; doi:10.1126/stke.2952005pe39
- Elbashir SM, et al. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498; doi:10.1038/35078107
- Fire A, Xu S, Montgomery MK, et al. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806-811; doi: 10.1038/35888
- Krishnaswamy S. The transition of prothrombin to thrombin (2013) J Thromb Haemost11 Suppl 1:265-276; doi:10.1111/jth.12217
- Negrier C, Shima M, Hoffman M (2019) The central role of thrombin in bleeding disorders. Blood Rev 38:100582; doi:10.1016/j.blre.2019.05.006
- Asakura H, et al. (1994) Study of the balance between coagulation and fibrinolysis in disseminated intravascular coagulation using molecular markers. Blood Coagulation & Fibrinolysis 5:829-832; doi: 10.1097/00001721-199410000-00022
- YourGenome: What is gene expression? (2016) Creative Commons 4.0 CC-BY license [online] Accessed 20 June 2022.
- Machin N, Ragni MV (2018) An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med 9:135-140; doi:10.2147/JBM.S159297
- Wang HW et al. (2009) Structural insights into RNA processing by the human RISC-loading complex. Nat Struct Mol Biol 16:1148–1153; doi: 10.1038/nsmb.1673
- Machin N, Ragni MV (2018) An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med 9:135-140; doi:10.2147/JBM.S159297
- Sehgal A, et al. (2015) An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med 21:492-497; doi: 10.1038/nm.3847
- Springer AD, Dowdy SF (2018) GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther 28:109-118; doi: 10.1089/nat.2018.0736
- Perrin GQ, Herzog RW, Markusic DM (2019) Update on clinical gene therapy for hemophilia. Blood 133:407-414; doi: 10.1182/blood-2018-07-820720
- Gale AJ (2011) Continuing education course #2: current understanding of hemostasis. Toxicol Pathol. 39:273-280; doi: 10.1177/0192623310389474