Hemophilia: Our Approach to New Therapeutics
People living with hemophilia are looking for treatments that last longer and make life more predictable and manageable. That’s why scientists are investigating three different approaches to preventing bleeds in people with missing or insufficient blood-clotting factors:
- Protein engineering: In clinical studies, we’re investigating a new class of factor VIII replacement therapy. It's designed to sustain high protection from bleeds in people with hemophilia A, with factor levels in the normal to near-normal range for most of the week.
- RNA technology: Our goal is to help people live more active lives and think about their treatments less. That’s why we’re working to provide extended protection with minimal treatment burden. We’re studying a treatment that’s based on small interfering RNA (siRNA). It’s designed to rebalance the missing blood-clotting factors in people with hemophilia A and B, with or without inhibitors (antibodies that attack replacement factors), potentially offering prophylaxis with as few as 6 injections per year.
- Gene therapy: Researchers are exploring ways to correct the single DNA mutation that prevents patients from producing enough blood-clotting factor.
1. Factor replacement therapies: protein engineering at work
Without enough factor VIII or IX, the body can’t produce enough thrombin to activate clotting.1,2 To solve this problem, many people are treated with replacement factors, given by intravenous (IV) injection. These replacement factors circulate in the blood until they’re either metabolized or activated to help stop bleeding.
But factor proteins are cleared from the body so quickly that many people with hemophilia A need to inject replacement factor several times a week.
Extended half-life factor replacement therapies keep factor VIII or IX proteins circulating longer. This is possible thanks to earlier innovations in protein engineering. To protect the replacement factors from being metabolized too quickly, scientists designed them to “hitch a ride” on proteins that are recycled more slowly.
Breaking the 19-hour limit
But there’s a limit: clotting factor VIII can only circulate in the blood for between 15 and 19 hours. That’s because factor VIII usually travels with a companion protein called von Willebrand factor (vWF). vWF protects factor VIII from being broken down3,4but every 15-19 hours, half of it gets cleared from the blood, taking the factor along with it.
The challenge for scientists: to develop a factor VIII replacement therapy that keeps these clotting proteins circulating for longer.
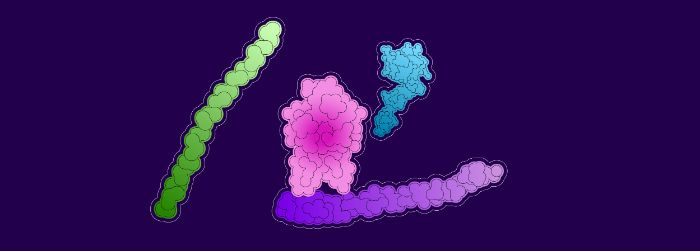
Von Willebrand factor (purple) protects factor VIII (pink) from being broken down by certain proteins (green and blue).
An alternative to von Willebrand Factor?
We’ve been developing a replacement factor VIII that enjoys the kind of protection vWF gives, without getting caught up in the vWF recycling pathway.5
- A small fragment of vWF is attached to the replacement factor as a decoy, designed to trick the body’s own vWF into ignoring it. A “linker” molecule releases the decoy when the factor is needed for clotting.
- Another molecule, XTEN®, acts as a shield, fending off other proteins to protect the replacement factor from being metabolized. An antibody domain helps the factor stay in circulation longer.
The ambition of this novel design, which is still being investigated, is to sustain factor VIII levels in the normal to near-normal range for more than half the week.
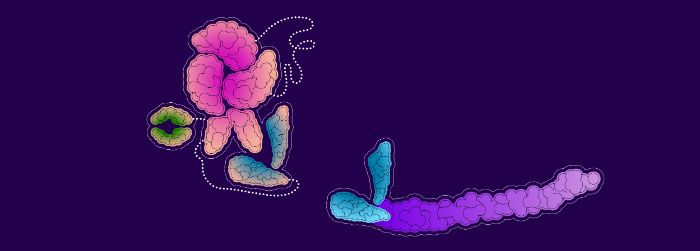
When replacement factor VIII (yellow) is bound to a vWF domain (blue), the vWF circulating in the blood (blue and gray) cannot bind to it.
2. RNA technology and hemophilia
mRNA has gained widespread attention for its role in vaccines, but other RNA technologies are also opening up new possibilities for healthcare. For example, our teams aim to improve blood clotting in people with hemophilia by using small interfering RNA (siRNA).6,7,8 It can be designed to help slow the body's manufacture of a protein called antithrombin-3.
Antithrombin is a protein that helps keep blood flowing by limiting the amount of thrombin the body generates. People with less antithrombin in their blood generate more thrombin than others. Recently, scientists discovered that having lower levels of antithrombin-3 restores some thrombin production in people with hemophilia, making clotting more effective.9,10,11
How can we slow down antithrombin-3 production?
The body uses mRNA templates to produce proteins, such as antithrombin-3.12 Without those mRNA templates, the proteins can’t be made. Our teams are studying an siRNA /enzyme complex that intercepts mRNA templates for antithrombin-3 and destroys enough of them to slow down protein production.13,14 That means antithrombin-3 levels could potentially remain low, leaving more thrombin available to initiate blood clotting.
This is the idea behind a potential prophylactic treatment for people with hemophilia A or B, with or without inhibitors.15,16,17 This type of treatment may result in even fewer treatment cycles for the patient.
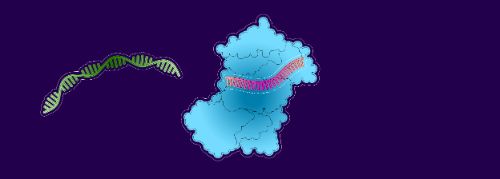
1. The siRNA lures an mRNA molecule on its way to deliver antithrombin-3 templates to a protein-building factory
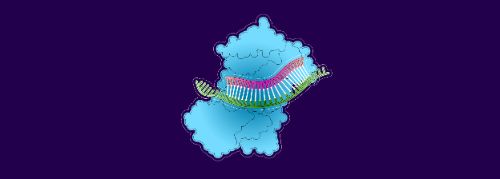
2. The mRNA binds to the siRNA
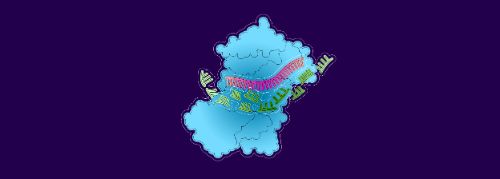
3. Enzymes break down the mRNA and clear it…
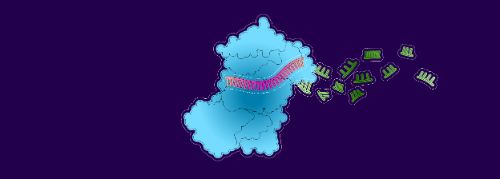
4. leaving the siRNA strand free to attract another mRNA
3. Gene therapies
In hemophilia research, the goal of gene therapy is to cure the disease by correcting the single DNA mutation that prevents patients from manufacturing enough factor VIII or IX. We're exploring experimental approaches to gene therapy that we hope will one day improve the lives of people with hemophilia and their families.18,19
This article refers to therapies that are being investigated in research and development programs. The safety and efficacy of the investigational medicines described here have not been evaluated by any regulatory authority.
Explore more

For Hemophilia Heroes
Restoring Balance in the Blood
Rare Blood Disorders R&D
References
- Siegemund T, et al. (2003) Thrombin generation in severe haemophilia A and B: the endogenous thrombin potential in platelet-rich plasma. Thromb Haemost 90:781-786; doi:10.1160/TH03-01-0027
- Brummel-Ziedins KE, et al. (2009) Thrombin generation and bleeding in haemophilia A. Haemophilia 15:1118-1125; doi:10.1111/j.1365-2516.2009.01994
- Orlova NA, et al. (2013) Blood Clotting Factor VIII: From Evolution to Therapy. Acta Naturae 5:19-39
- Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124(9):1412-1425. doi:10.1182/blood-2014-05-378638
- Seth Chhabra E, Liu T, Kulman J, et al. (2020) BIVV001, a new class of factor VIII replacement for hemophilia A that is independent of von Willebrand factor in primates and mice. Blood 135:1484-1496; doi: 10.1182/blood.2019001292
- Bartz S, Jackson AL (2005) How will RNAi facilitate drug development? Sci STKE: Signal Transduction Knowledge Environment. 295:pe39; doi:10.1126/stke.2952005pe39
- Elbashir SM, et al. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498; doi:10.1038/35078107
- Fire A, Xu S, Montgomery MK, et al. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806-811; doi: 10.1038/35888
- Krishnaswamy S. The transition of prothrombin to thrombin (2013) J Thromb Haemost11 Suppl 1:265-276; doi:10.1111/jth.12217
- Negrier C, Shima M, Hoffman M (2019) The central role of thrombin in bleeding disorders. Blood Rev 38:100582; doi:10.1016/j.blre.2019.05.006
- Asakura H, et al. (1994) Study of the balance between coagulation and fibrinolysis in disseminated intravascular coagulation using molecular markers. Blood Coagulation & Fibrinolysis 5:829-832; doi: 10.1097/00001721-199410000-00022
- YourGenome: What is gene expression? (2016) Creative Commons 4.0 CC-BY license [online] Accessed 20 June 2022.
- Machin N, Ragni MV (2018) An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med 9:135-140; doi:10.2147/JBM.S159297
- Wang HW et al. (2009) Structural insights into RNA processing by the human RISC-loading complex. Nat Struct Mol Biol 16:1148–1153; doi: 10.1038/nsmb.1673
- Machin N, Ragni MV (2018) An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med 9:135-140; doi:10.2147/JBM.S159297
- Sehgal A, et al. (2015) An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med 21:492-497; doi: 10.1038/nm.3847
- Springer AD, Dowdy SF (2018) GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther 28:109-118; doi: 10.1089/nat.2018.0736
- Perrin GQ, Herzog RW, Markusic DM (2019) Update on clinical gene therapy for hemophilia. Blood 133:407-414; doi: 10.1182/blood-2018-07-820720
- Gale AJ (2011) Continuing education course #2: current understanding of hemostasis. Toxicol Pathol. 39:273-280; doi: 10.1177/0192623310389474